Sickle Cell Anemia Pain
What is sickle cell anemia disease
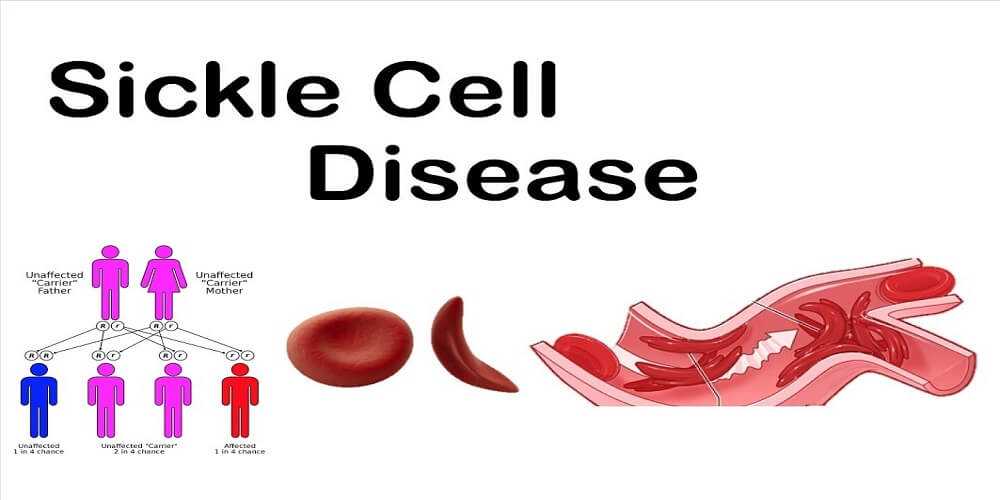
Sickle Cell Anemia (SCA) effects over 70,000 Americans. Each year 1000 additional children are born with SCA. If a person is of North African Sub-Saharan ancestry they will have a 1/500 chance of being born with SCA.

Other ethnic groups are at risk too. Nearly any ethnic group that has its’ origins from the North African region can be expected to be at risk for SCA (some Hispanic people are at risk for SCA too).
The diagnosis of SCA is easily established by looking at a smear of red blood cells and finding the presence of “sickled” red blood cells. Genetic testing is now available that can predict the likelihood that parents in a high risk ethnic group carry the gene for the abnormal hemoglobin.
Ironically, the person with SCA is at less risk of developing Malaria (a blood infection that is endemic in the same area where SCA predominates in Africa). So it is that the disadvantage of having SCA gives a selective survival advantage to those living in a high Malaria region.
This posting is going to focus on Sickle Cell Anemia pain, its’ mechanisms, and treatment. A more thorough discussion of SCA can be obtained by reading the source document links at the end of this article.
Anatomy of Sickle Cell Anemia
The human body is nourished by blood and instructed by nerves. The blood system is the “highway” through which oxygen, nutrients, and waste products are moved. It has been rightly said, “the life is in the blood…” and Sickle Cell Anemia demonstrates this principle.
Imagine you are driving on a highway into a major city and an accident occurs. The cars, trucks, and debris block traffic into the city. The traffic is all “jammed up” for miles. The tractor trailers full of food for the restaurants in the city are stuck on the highway.
People needing to get home and rest after a hard day’s work are caught in the traffic jam. People rested and wanting to get into the city are stuck. How long could any major city survive with the roads leading into it “jammed up?”
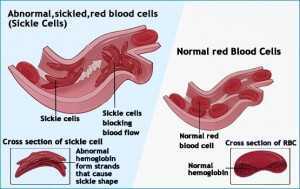
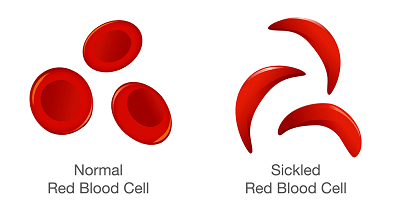
Well, that is what happens in Sickle Cell Anemia. The red blood cells of Sickle Cell Anemia have a tendency to become stiff, change in shape from a soft “bagel” to one of a rigid “sickle.” In the next section I will explain what rigid blood cells mean for biologic function.
Pathophysiology of Sickle Cell Anemia
Every organ system of the human body requires good blood flow. Without it the function of any given system starves. Depending on the requirements for blood flow and nutrients, the symptoms that develop may occur slowly or very quickly if the blood flow is reduced. For instance, the brain utilizes 25% of all the oxygen in the blood. Deprive it of oxygen and in short order (5 minutes or so) irreversible injury occurs.

There are no exceptions to this rule only differing requirements. Some organ systems require less oxygen so that their injury from reduced blood flow is less dramatic.
Oxygen is primarily carried in the blood stream “piggy-back” style on a molecule called hemoglobin. Hemoglobin is located in red blood cells. So it can be said that red blood cells carry oxygen.
The red blood cells are like a never ending “freight train” full of oxygen. Each red blood cell (RBC) is a “box car” full of oxygen. The blood stream takes the RBC through smaller and smaller tubes (blood vessels) to a place where the RBC has to squeeze down and bend to get into position to off load its’ cargo…oxygen.
It is in these tight areas, called capillaries, that the SCA RBC has the most trouble. Because it is stiff and shaped like a “sickle” it “slices up” the interior of the capillary. The debris from the “slicing” as well as the stiff SCA RBC cause a “traffic jam.” Blood flow comes to a halt.
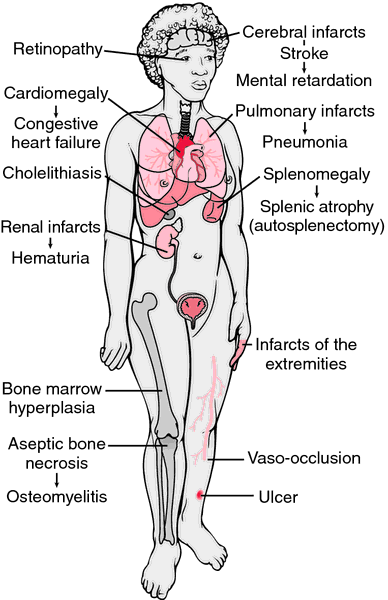
The organs that are the most sensitive to the damaging effects of this process are the systems that require the most blood flow. You could probably predict which are injured the most. In the next section we will introduce the nerve injury by SCA. This injury also results in a severe, chronic pain syndrome for many people with SCA.
The Mechanism for the Chronic Pain of Sickle Cell Anemia
The medical profession has previously focused on the acute pain episodes of patients with SCA. Recent reviews of pain management in the pediatric population reveals that 77% of all children treated for acute pain syndromes related to SCA are undertreated.
The chronic pain syndromes of people with SCA have not been well understood. Whereas, acute pain in SCA usually reveals an identifiable source, the mechanism of generation of chronic pain in SCA has been elusive.
Because of the lack of easily identifiable mechanisms for chronic pain in people with SCA, as well as the general antipathy to using opiates in high doses for chronic pain, the SCA patient with chronic pain is usually undertreated.
A recent report that examines neuropathic pain (the hallmarks of which are burning pain, an “exaggerated” response to a usually painful stimulus called hyperalgesia, and a painful response to a non-painful stimulus called allodynia) revealed that 90% of adult SCA patients reported this pattern.
Furthermore, 57% of the same group reported the pain as being continuous. An examination of their pain treatment regimen revealed that no patient was receiving adequate pain management for their neuropathic pain.
The pre-occupation with acute pain, and the need to demonstrate some diagnostic finding before initiating effective pain management, leaves the SCA patient with chronic pain insufficiently treated. These patients often roam from emergency room to emergency room…doctor to doctor…and become labeled as “drug seekers.” More recently, with the available data on neuropathic pain mechanisms, perhaps the more accurate label should be “pain relief seeking” behavior.
Neuropathic pain in SCA patients seems to be mediated by a pain receptor that enhances transmission of pain called the NMDA receptor. This receptor is located in various regions of the nervous system and is stimulated by 2 particular substances found in abundance in SCA patients (PKC-Protein Kinase C and CaMKII-Calcium/Calmodulin dependent protein kinase II).
It is becoming clearer that the chronic pain syndrome in SCA patients is a reality. Perhaps the “tide is turning” in favor of more aggressive treatment of the chronic pain syndrome in SCA patients.
Treatment for Sickle Cell Anemia
The treatment of SCA is separated into 4 components:
1) Prevention of Acute SCA Episodes:
- Avoid Dehydration
- Avoid Smoking and Drinking Alcohol
- Avoidance of Infection
- Pneumococcal Vaccination
- Routine Vaccinations
- Close medical follow-up preferably with a Hematologist (blood specialist)
- Stress reduction
- No illicit drug use
2) Treatment of Acute SCA Episodes:
- Immediate medical evaluation
- Rehydration
- Pain management
- Identify all complications
- Treat infections
- Possible Hospitalization
3) Treatment of Chronic Complications:
Nearly any organ system can be effected by SCA. A regular thorough evaluation by the same health care practitioner is optimal for providing continuity of care. Survival rates for SCA people are highest when they have continuity for their care so that early complications may be intercepted.
4) Treatment of Chronic Pain:
This has been problematic for the SCA patient. For more insight on this topic see my posts on “Chronic Pain and Addiction” as well as “Pain Myhts.”

The source articles at the end of this post will also give more information on this topic.
Survival rate of Sickle Cell Anemia
Sickle Cell Anemia impacts the overall life expectancy of an individual. Depending on the type of Sickle Cell Anemia, women have a median life expectancy of 48-68 years. The male life expectancy range is 42-60 years.
Much depends on the availability of comprehensive medical care and the diligence of the person with Sickle Cell Anemia to avoid “triggers” for complications.
Summary Comments
I have attempted to explain sickle cell anemia pain from the neuropathic point of view. This form of chronic pain has been previously poorly understood by most practicing clinicians for the Sickle Cell Anemia patient.
I hope you have enjoyed this post. If you have additional questions please contact me at below. I would love to hear from you.
I wish you much joy and good health.